GRaSP
GRaSP is a residue centric method to predict ligand biding site residues. It is based on a supervised learning strategy that models the residue environment as a graph at the atomic level.
GRaSP is a residue centric method to predict ligand biding site residues. It is based on a supervised learning strategy that models the residue environment as a graph at the atomic level.
For each residue, physicochemical and topological properties of its atoms and non-covalent interactions are modeled as a graph which, in turn, is encoded as a feature vector. A set of feature vectors is the input for the machine learning predictor.

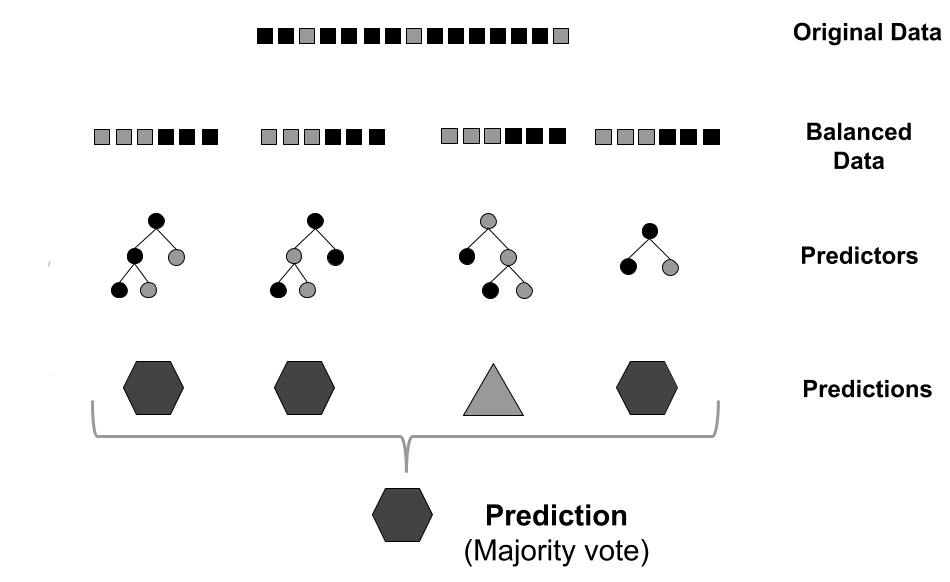
GRaSP uses the residues environment, modeled as feature vectors, as input to a machine learning strategy. The prediction is performed using a balancing strategy to reduce the imbalanced distribution of classes.